Features
Data access
Dataset is an abstraction of the local file system. Users can add their local dirs into this system then load data that under these dirs in a convenient way.
XenonPy also uses this system to provide some built-in data.
Currently, two sets of element-level property data are available out-of-the-box (elements and elements_completed (imputed version of elements)).
These data were collected from mendeleev, pymatgen, CRC Hand Book and Magpie.
elements contains 74 element-level properties of 118 elements. Their missing values
were statistically imputed by performing the multiple imputation method [1] and stored as elements_completed.
Because of the statistical unreliability of the imputation for a subset of properties and heavier atoms that contains many missing values in elements,
the elements_completed data set provides only 58 properties of 94 elements (from H to Pu). The following table shows the currently available elemental information.
feature |
description |
|
Period in the periodic table |
|
Number of protons found in the nucleus of an atom |
|
Atom number in mendeleev’s periodic table |
|
Atomic radius |
|
Atomic radius by Rahm et al |
|
Atomic volume |
|
The mass of an atom |
|
Atom volume in ICSD database |
|
Physical dimension of unit cells in a crystal lattice |
|
Van der Waals radius |
|
Van der Waals radius according to Alvarez |
|
Van der Waals radius according to Batsanov |
|
Van der Waals radius according to Bondi |
|
Van der Waals radius from the DREIDING FF |
|
Van der Waals radius from the MM3 FF |
|
Van der Waals radius according to Rowland and Taylor |
|
Van der Waals radius according to Truhlar |
|
Van der Waals radius from the UFF |
|
Covalent radius by Bragg |
|
Covalent radius by Cerdero et al |
|
Single bond covalent radius by Pyykko et al |
|
Double bond covalent radius by Pyykko et al |
|
Triple bond covalent radius by Pyykko et al |
|
Covalent radius by Slater |
|
C_6 dispersion coefficient in a.u |
|
C_6 dispersion coefficient in a.u |
|
Density at 295K |
|
Proton affinity |
|
Dipole polarizability |
|
Electron affinity |
|
Tendency of an atom to attract a shared pair of electrons |
|
Allen’s scale of electronegativity |
|
Ghosh’s scale of electronegativity |
|
Pauling’s scale of electronegativity |
|
DFT bandgap energy of T=0K ground state |
|
DFT energy per atom (raw VASP value) of T=0K ground state |
|
Estimated BCC lattice parameter based on the DFT volume |
|
Estimated FCC lattice parameter based on the DFT volume |
|
DFT magnetic momenet of T=0K ground state |
|
DFT volume per atom of T=0K ground state |
|
Herfindahl−Hirschman Index (HHI) production values |
|
Herfindahl−Hirschman Index (HHI) reserves values |
|
Specific heat at 20oC |
|
Gas basicity |
|
First ionisation energy |
|
Fusion heat |
|
Heat of formation |
|
Mass specific heat capacity |
|
Molar specific heat capacity |
|
Evaporation heat |
|
Coefficient of linear expansion |
|
Boiling temperature |
|
Brinell Hardness Number |
|
Bulk modulus |
|
Melting point |
|
Single-bond metallic radius |
|
Metallic radius with 12 nearest neighbors |
|
Thermal conductivity at 25 C |
|
Speed of sound |
|
Value of Vickers hardness test |
|
Ability to form instantaneous dipoles |
|
Young’s modulus |
|
Poisson’s ratio |
|
Molar volume |
|
Total unfilled electron |
|
Total valance electron |
|
Unfilled electron in d shell |
|
Valance electron in d shell |
|
Unfilled electron in f shell |
|
Valance electron in f shell |
|
Unfilled electron in p shell |
|
Valance electron in p shell |
|
Unfilled electron in s shell |
|
Valance electron in s shell |
For more details on this system, see Data access.
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/dataset_and_preset.ipynb to get a runnable script.
Descriptor calculation
Compositional descriptors
XenonPy can calculate 290 compositional features for a given chemical composition.
This calculation uses the information of the 58 element-level property data recorded in elements_completed.
For example, let us consider a binary compound, \(A_{w_A}B_{w_B}\), whose element-level features are denoted by \(f_{A,i}\) and \(f_{B,i} (i = 1, …, 58)\). Then, the 290 compositional descriptors are calculated: for \(i = 1, …, 58\),
Weighted average (abbr: ave): \(f_{ave, i} = w_{A}^* f_{A,i} + w_{B}^* f_{B,i}\),
Weighted variance (abbr: var): \(f_{var, i} = w_{A}^* (f_{A,i} - f_{ave, i})^2 + w_{B}^* (f_{B,i} - f_{ave, i})^2\),
Geometric mean (abbr: gmean): \(f_{gmean, i} = \sqrt[w_A + w_B]{f_{A,i}^{w_A} * f_{V,i}^{w_B}}\),
Harmonic mean (abbr: hmean): \(f_{hmean, i} = \frac{w_A +w_B}{\frac{1}{f_{A,i}}*w_A + \frac{1}{f_{B,i}}*w_B}\),
Max-pooling (abbr: max): \(f_{max, i} = max{f_{A,i}, f_{B,i}}\),
Min-pooling (abbr: min): \(f_{min, i} = min{f_{A,i}, f_{B,i}}\),
Weighted sum (abbr: sum): \(f_{sum, i} = w_{A} f_{A,i} + w_{B} f_{B,i}\),
where \(w_{A}^*\) and \(w_{B}^*\) denote the normalized composition summing up to one.
By using compositional descriptors, we have succeeded in predicting the composition of quasicrystals [2].
Structural descriptors
Currently, XenonPy implements RDF (radial distribution function) and OFM (orbital field matrix [3]) descriptors of crystalline structures. We also provide a compatible API to use the structural descriptors of matminer. You may check the summary table of featurizers in matminer here.
RDKit descriptors
XenonPy also supports molecular descriptors available in the RDKit python package, including 10 sets of fingerprints, each contains corresponding options.
The tutorial at Descriptor calculation demonstrates how to calculate descriptors using XenonPy.descriptor classes.
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/calculate_descriptors.ipynb to get a runnable script.
Visualization of descriptor-property relationships
Descriptors on a set of given materials could be displayed on a heatmap plot in order to facilitate the understanding of overall patterns in relation to their properties. The following figure shows an example:
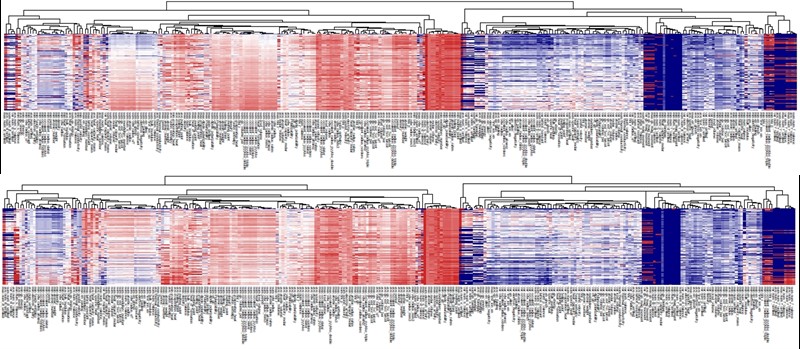
Heatmap of 290 compositional descriptors of 69,640 compounds in Materials Project (upper: volume Å3, lower: density g/cm3 ).
In the heatmap of the descriptor matrix, the 69,640 materials are arranged from the top to bottom by the increasing order of formation energies. Plotting the descriptor-property relationships in this way, we could visually recognize which descriptors are relevant or irrelevant to the prediction of formation energies. Relevant descriptors, which are linearly or nonlinearly dependent to formation energies, might exhibit certain patterns from top to bottom in the heatmap. For example, a monotonically decrease or increase pattern would appear in a linearly dependent descriptor. On the other hand, irrelevant descriptors might exhibit no specific patterns.
See the tutorial for visualization of descriptor-property relationships at Visualization.
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/visualization.ipynb to get a runnable script.
XenonPy.MDL
WARNING: This subsection’s information is out-dated and the details are no long true. Currently XenonPy.MDL is undergoing indefinite maintenance. Please refer to the github README page for the latest update.
XenonPy.MDL is a library of pre-trained models that were obtained by feeding diverse materials data on structure-property relationships into neural networks and some other supervised learning algorithms. The current release (version 0.1.0.beta) contains more than 140,000 models (include private models) on physical, chemical, electronic, thermodynamic, or mechanical properties of small organic molecules (15 properties), polymers/polymer composites (18), and inorganic compounds (12). Pre-trained neural networks are distributed as either the R (MXNet) or Python (PyTorch) model objects. Detailed information about XenonPy.MDL, such as a list of models, properties, source data used for training, and so on, are prepared in this paper [3].
The following lists contain the information of current available pre-trained models and properties.
id |
name |
description |
|---|---|---|
|
Stable inorganic compounds
in materials project (MP)
|
Models in this set are trained on ~20,000 stable inorganic
compounds selected from the materials project.
|
|
All inorganic compounds
in materials project (MP)
|
Models in this set are trained on ~70,000 inorganic compounds
selected from the materials project.
|
|
QM9 Dataset from
Quantum-Machine website
|
Quantum-Machine project can be access
|
|
PHYSPROP Dataset |
PHYSPROP database contains chemical structures,
names and physical properties for over 41,000 chemicals.
|
|
Jean-Claude Bradley Open
Melting Point Dataset
|
Jean-Claude Bradley’s dataset of Open Melting Points.
|
|
Polymer Genome Dataset (PG)
|
Polymer Genome is an informatics platform for polymer property
prediction and design using machine learning.
It can be accessed via https://www.polymergenome.org/.
|
name |
system |
querying name |
Melting Temperature |
Organic Polymer |
organic.polymer.melting_temperature |
Ionization Energy |
Organic Polymer |
organic.polymer.ionization_energy |
Ionic Dielectric Constant |
Organic Polymer |
organic.polymer.ionic_dielectric_constant |
Hildebrand Solubility Parameter |
Organic Polymer |
organic.polymer.hildebrand_solubility_parameter |
Glass Transition Temperature |
Organic Polymer |
organic.polymer.glass_transition_temperature |
Molar Volume |
Organic Polymer |
organic.polymer.molar_volume |
Electron Affinity |
Organic Polymer |
organic.polymer.electron_affinity |
Dielectric Constant |
Organic Polymer |
organic.polymer.dielectric_constant |
Density |
Organic Polymer |
organic.polymer.density |
Cohesive Energy |
Organic Polymer |
organic.polymer.cohesive_energy |
Bandgap |
Organic Polymer |
organic.polymer.bandgap |
Atomization Energy |
Organic Polymer |
organic.polymer.atomization_energy |
Refractive Index |
Organic Polymer |
organic.polymer.refractive_index |
Molar Heat Capacity |
Organic Polymer |
organic.polymer.molar_heat_capacity |
Electronic Dielectric Constant |
Organic Polymer |
organic.polymer.electronic_dielectric_constant |
U0 Hartree |
Organic Nonpolymer |
organic.nonpolymer.u0_hartree |
R2 Bohr2 |
Organic Nonpolymer |
organic.nonpolymer.r2_bohr2 |
Mu Debye |
Organic Nonpolymer |
organic.nonpolymer.mu_debye |
Lumo Hartree |
Organic Nonpolymer |
organic.nonpolymer.lumo_hartree |
Homo Hartree |
Organic Nonpolymer |
organic.nonpolymer.homo_hartree |
Gap Hartree |
Organic Nonpolymer |
organic.nonpolymer.gap_hartree |
Alpha Bohr3 |
Organic Nonpolymer |
organic.nonpolymer.alpha_bohr3 |
U Hartree |
Organic Nonpolymer |
organic.nonpolymer.u_hartree |
Zpve Hartree |
Organic Nonpolymer |
organic.nonpolymer.zpve_hartree |
Bp |
Organic Nonpolymer |
organic.nonpolymer.bp |
Cv Calmol-1K-1 |
Organic Nonpolymer |
organic.nonpolymer.cv_calmol-1k-1 |
Tm |
Organic Nonpolymer |
organic.nonpolymer.tm |
G Hartree |
Organic Nonpolymer |
organic.nonpolymer.g_hartree |
H Hartree |
Organic Nonpolymer |
organic.nonpolymer.h_hartree |
Density |
Inorganic Crystal |
inorganic.crystal.density |
Volume |
Inorganic Crystal |
inorganic.crystal.volume |
Refractive Index |
Inorganic Crystal |
inorganic.crystal.refractive_index |
Band Gap |
Inorganic Crystal |
inorganic.crystal.band_gap |
Dielectric Const Electron |
Inorganic Crystal |
inorganic.crystal.dielectric_const_elec |
Fermi Energy |
Inorganic Crystal |
inorganic.crystal.efermi |
Total Magnetization |
Inorganic Crystal |
inorganic.crystal.total_magnetization |
Dielectric Const Total |
Inorganic Crystal |
inorganic.crystal.dielectric_const_total |
Final Energy Per Atom |
Inorganic Crystal |
inorganic.crystal.final_energy_per_atom |
Formation Energy Per Atom |
Inorganic Crystal |
inorganic.crystal.formation_energy_per_atom |
XenonPy.MDL provides a rich set of APIs to give users the abilities to interact with the pre-trained model database. Through the APIs, users can search for a specific subset of models by keywords and download them via HTTP. The tutorial at Pre-trained Model Library illustrates how to interact with the database in XenonPy (via the API querying).
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/pre-trained_model_library.ipynb to get a runnable script.
Transfer learning
Transfer learning has become one of the basic techniques in machine learning that covers a broad range of algorithms for which a model trained for one task is re-purposed to another related task [4] [5]. In general, the need of transfer learning occurs when there is a limited supply of training data for a specific task, yet data supply for related task is sufficient. This situation occurs in many materials science applications as described in [6] [7].
XenonPy offers a simple-to-use toolchain to seamlessly perform transfer learning with the given pre-trained models. Given a target property, by using the transfer learning module of XenonPy, a source model can be treated as a generator of machine learning acquired descriptors, so-called the neural descriptors, as demonstrated in [3].
See tutorial at Transfer Learning to learn how to perform frozen feature transfer learning in XenonPy.
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/transfer_learning.ipynb to get a runnable script.
Inverse design
Inverse molecular design is an important research subject in materials science that aims to create new chemical structures with desired properties computationally. XenonPy offers a Bayesian molecular design algorithm based on [8] that includes a SMILES generator based on N-gram model, likelihood calculator, and a sequential Monte Carlo algorithm for sampling the posterior distribution of molecules with properties specified by the likelihood function. Details of this algorithm, which is called iQSPR, can be found in [9]. An example of using iQSPR to search for high thermal conductivity polymer can be found in [10].
See tutorial at XenonPy-iQSPR tutorial to learn how to perform inverse molecular design using iQSPR in XenonPy.
Access https://github.com/yoshida-lab/XenonPy/blob/master/samples/iQSPR.ipynb to get a runnable script.
Reference